Púrpura de Henoch-Schönlein
| Púrpura de Henoch-Schönlein | |
|---|---|
| Clasificación y recursos externos | |

Púrpura típica en las extremidades inferiores y nalgas
|
|
| CIE-10 | D69,0 (ILDS D69.010) |
| CIE-9 | 287.0 |
| DiseasesDB | 5705 |
| MedlinePlus | 000425 |
| eMedicine | Derm/177 EMERG/767 EMERG/845 PED/3020 |
| Malla | D011695 |
Púrpura de Henoch-Schönlein (HSP, también conocido como púrpura anafilactoide,[1] púrpura de Henoch-Schönlein,[1] y Púrpura de Schönlein-Henoch)[1] es una enfermedad de la piel y otros órganos que más comúnmente afecta a los niños. En la piel, la enfermedad causa púrpura palpable (pequeñas hemorragias); a menudo con empalme y dolor abdominal. Con riñón participación, puede haber una pérdida de pequeñas cantidades de sangre y proteína en la orina, pero esto generalmente pasa desapercibida; en una pequeña proporción de los casos, la participación del riñón se procede a enfermedad renal crónica. HSP es a menudo precedida por una infección, como una infección de la garganta.
HSP es un sistémico vasculitis (inflamación de los vasos sanguíneos) y se caracteriza por la deposición de complejos inmunes que contiene el anticuerpo IgA; se desconoce la causa exacta de este fenómeno. Generalmente se resuelve dentro de varias semanas y no requiere ningún tratamiento aparte de control de los síntomas, pero puede recaer en un tercio de los casos y causar daño renal irreversible en aproximadamente uno de cada 100 casos.
Contenido
- 1 Signos y síntomas
- 2 Fisiopatología
- 3 Diagnóstico
- 3.1 Clasificación
- 4 Tratamiento
- 5 Pronóstico
- 5.1 Implicación del riñón
- 6 Epidemiología
- 7 Historia
- 8 Véase también
- 9 Referencias
Signos y síntomas


Púrpura, artritis y dolor abdominal son conocidos como la "tríada clásica" de la púrpura de Henoch-Schönlein.[2] Púrpura ocurren en todos los casos, dolores articulares y artritis en un 80% y el dolor abdominal en el 62%. Algunos incluyen hemorragia gastrointestinal como un cuarto criterio; Esto ocurre en el 33% de los casos, a veces, pero no necesariamente siempre, debido a invaginación intestinal.[3] La púrpura típicamente aparecen en las piernas y glúteos, pero también puede ser visto en los brazos, cara y tronco. El dolor abdominal es de tipo cólico en carácter y puede acompañarse de náuseas, vómitos, estreñimiento o diarrea. Puede haber sangre o moco en las heces.[4] Las articulaciones implicadas tienden a ser los tobillos, rodillas, y codos, pero la artritis en las manos y los pies es posible; la artritis es no erosiva y por lo tanto no causa ninguna deformidad permanente.[2] Cuarenta por ciento tienen evidencia de riñón participación, principalmente en forma de hematuria (sangre en la orina), pero sólo una cuarta parte tendrá esto en cantidades suficientes para ser visibles sin pruebas de laboratorio.[3] Problemas en otros órganos, tales como la sistema nervioso central (cerebro y médula espinal) y pulmones puede ocurrir, pero es mucho menos común que en la piel, del intestino y los riñones.[5]
Del 40% de los pacientes que desarrollan la implicación renal, casi todos han evidencia (visible o en Análisis de orina) de la sangre en la orina. Más de la mitad también tienen proteinuria (proteína en la orina), que en un octavo es lo suficientemente grave como para causar síndrome nefrótico (generalizadas hinchazón debido al contenido de baja proteína de la sangre). Mientras que las anormalidades en el análisis de orina pueden continuar por mucho tiempo, sólo el 1% de todos los pacientes HSP desarrollar enfermedad renal crónica.[5] Hipertensión (presión arterial alta) puede ocurrir. Alta y pérdida de la proteína de la sangre la presión, así como las características en biopsia del riñón si se realiza, se puede predecir la progresión a la enfermedad renal avanzada. Los adultos son más propensos que los niños desarrollen la enfermedad renal avanzada.[5][6]
Fisiopatología
Púrpura de Henoch-Schönlein es una vasculitis de vasos pequeños en que los complejos de inmunoglobulina A (IgA) y componente del complemento 3 (C3) se depositan en las arteriolas, capilares y vénulas. Como con Nefropatía por IgA, los niveles séricos de IgA son altos en HSP y hay resultados idénticos en biopsia renal; Sin embargo, la nefropatía por IgA tiene una predilección para los adultos jóvenes mientras HSP es más predominante entre los niños. Además, la nefropatía por IgA normalmente sólo afecta a los riñones mientras HSP es una enfermedad sistémica. HSP implica la piel y los tejidos conectivos, escroto, articulaciones, tracto gastrointestinal y los riñones.[7]
Diagnóstico
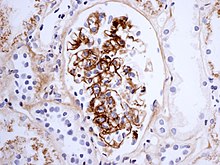
El diagnóstico se basa en la combinación de los síntomas, como muy pocas otras enfermedades causan los mismos síntomas juntos. Exámenes de sangre puede mostrar elevadas creatinina y urea niveles (en la implicación del riñón), criados IgA niveles (en aproximadamente el 50%[7]) y criado Proteína C reactiva (CRP) o tasa de sedimentación eritrocítica Resultados (ESR); Ninguno es específico para la púrpura de Henoch-Schönlein. El plaqueta puede ser levantado y distingue de enfermedades donde las plaquetas bajas son la causa de la púrpura, tales como púrpura trombocitopénica idiopática y púrpura trombocitopénica trombótica.[2]
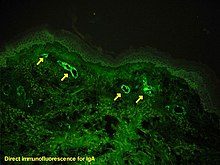
Si hay duda sobre la causa de las lesiones de piel, un biopsia de la piel puede realizarse para distinguir la púrpura de otras enfermedades que causan, tales como vasculitis debido a Crioglobulinemia; en microscopia las apariencias son de un vasculitis de la hipersensibilidad, y inmunofluorescencia demuestra IgA y C3 (una proteína de la sistema del complemento) en la pared del vaso sanguíneo.[2] Sin embargo, los niveles de complemento del suero total son normales.
Sobre la base de los síntomas, es posible distinguir HSP de vasculitis de la hipersensibilidad (HV). En una serie comparando 85 pacientes HSP con 93 pacientes HV, cinco síntomas fueron encontrados para ser indicativo de HSP: púrpura palpable, angina abdominal, hemorragia digestiva (no debido a la intussussception), hematuria y edad de menos de 20. La presencia de tres o más de estos indicadores tiene un 87% sensibilidad para la predicción de HSP.[8]
La biopsia de la riñón puede realizarse tanto para establecer el diagnóstico o para evaluar la severidad de la enfermedad renal ya sospecha. Los principales resultados de la biopsia del riñón son células crecientes y la deposición de Ig en el Mesangio (parte de la glomérulodonde se filtra la sangre), glóbulos blancosy el desarrollo de Crescent. Los cambios son indistinguibles de los observados en Nefropatía por IgA.[7]
HSP puede convertirse después de las infecciones con estreptococos (Β-hemolítico, grupo Lancefield A), hepatitis B, virus del herpes simple, parvovirus B19, Virus Coxsackie, adenovirus, Helicobacter pylori,[5] sarampión, paperas, rubéola, Mycoplasma y muchos otros.[7] Drogas vinculadas a HSP, generalmente como una reacción idiosincrásica, incluyen los antibióticos vancomicina y Cefuroxima, Inhibidores de ECA enalapril y captopril, agente antiinflamatorio Diclofenac, así como ranitidina y estreptoquinasa. Varias enfermedades se han divulgado para ser asociado de HSP, a menudo sin una relación causal. Sólo en alrededor del 35% de los casos HSP se remontan a cualquiera de estas causas.[7]
Se desconoce la causa exacta de HSP, pero la mayoría de sus características son debido a la deposición de anticuerpos anormales en la pared de los vasos sanguíneos, llevando a vasculitis. Estos anticuerpos son de la subclase IgA1 en polímeros; es incierto si la causa principal es la superproducción (en el tubo digestivo o la médula ósea) o disminución de la eliminación de IgA anormal de la circulación.[7] Se sospecha que las anormalidades en el IgA1 molécula puede proporcionar una explicación para su comportamiento anormal en la condición relacionada y HSP Nefropatía por IgA. Una de las características de IgA1 (y IgD) es la presencia de una joven de 18 aminoácido-tiempo "bisagra región" entre complemento-fijación de las regiones 1 y 2. De los aminoácidos, la mitad es prolina, mientras que los otros son principalmente serina y Treonina. La mayoría de las serinas y los threonines tienen azúcar elaborada cadenas, conectadas a través de oxígeno los átomos)O-glicosilación). Este proceso está pensado para estabilizar la molécula de IgA y hacerlo menos propenso a proteólisis. El primer azúcar siempre es N-acetil-galactosamina (GalNAc), seguido de otro galactoses y ácido siálico. En HSP y nefropatía por IgA, estas cadenas de azúcar parecen ser deficientes. No se conoce la razón exacta de estas anormalidades.[5][7]
Clasificación
Existen varios estándares para la definición de púrpura de Henoch-Schönlein, incluyendo los años 1990 Colegio estadounidense de Reumatología Clasificación (ACR)[9][10] y la Conferencia de consenso de Chapel Hill 1994 (CHCC).[11] Algunos han informado los criterios del ACR para ser más sensible que los de la CHCC.[12]
Clasificaciones más recientes, el 2006 Liga Europea contra el reumatismo (EULAR) y Sociedad de Reumatología pediátrica Clasificación (PReS), incluyen púrpura palpable como un criterio obligatorio, junto con al menos uno de los siguientes hallazgos: difuso dolor abdominal, la deposición de IgA predominante (confirmado por biopsia de la piel), artritis aguda en cualquier participación conjunta y renal (según lo evidenciado por la presencia de sangre o proteína en la orina).[13]
Tratamiento
Analgésicos puede ser necesaria para los dolores abdominales y conjuntos. Es incierto en cuanto a si HSP necesita tratamiento más allá de controlar los síntomas. Mayoría de los pacientes no reciben la terapia debido a la tasa alta recuperación espontánea. Esteroides generalmente se evitan.[5] Sin embargo, si se les temprano en el episodio de la enfermedad, puede acortar la duración de los síntomas, y dolor abdominal puede mejorar significativamente. Además, se puede reducir el riesgo de problemas renales graves.[14] Sin embargo, cierta evidencia sugiere que los esteroides no disminuye la probabilidad de desarrollar enfermedad renal a largo plazo.[15]
Evidencia de daño renal empeora normalmente provocaría una biopsia de riñón. Puede indicarse tratamiento sobre la base de la apariencia de la muestra de la biopsia; pueden utilizarse varios tratamientos, que van desde los esteroides orales a una combinación de anestesia intravenosa metilprednisolona (esteroide), ciclofosfamida y dipiridamol seguidos por la prednisona. Otros regímenes incluyen esteroides /azatioprinay los esteroides/ciclofosfamida (con o sin heparina y warfarina). Inmunoglobulina intravenosa (IgIV) se utiliza de vez en cuando.[7]
Pronóstico
En general el pronóstico es bueno en la mayoría de los pacientes, con un estudio que muestra recuperación que ocurren en el 94% y 89% de los niños y adultos, respectivamente (algunos que necesitaban tratamiento).[16] En niños menores de diez años, la condición se repite en alrededor de un tercio de todos los casos y generalmente dentro de los primeros cuatro meses después del ataque inicial.[3] La recurrencia es más común en adultos y niños mayores.[5]
Implicación del riñón
En los adultos, implicación del riñón progresa a enfermedad renal en etapa final Niños (ESRD) más a menudo que en. En una serie de UK de 37 pacientes, 10 (27%) desarrollaron enfermedad renal avanzada. Proteinuria, hipertensión en la presentación y características de la patología (cambios Crescent, fibrosis intersticial y atrofia tubular) predijo la progresión.[6] Aproximadamente el 20% de los niños que presentan características nefrótico o nefrítico experiencia larga permanente deterioro renal.[17]
Los resultados en biopsia renal correlación con la severidad de los síntomas: aquellos con hematuria asintomática pueden tener sólo proliferación mesangial focal mientras que aquellos con proteinuria pueden haber marcada proliferación celular o incluso creciente formación. El número de glomérulos Crescent es un factor pronóstico importante en determinar si el paciente desarrollará la enfermedad renal crónica.[5]
En ERT, algunos necesitan eventualmente hemodiálisis o equivalente de la terapia de reemplazo renal (TRR). Si un trasplante de riñón se encuentra a un paciente en RRT, la enfermedad volverá a repetirse en el injerto (riñón trasplantado) en alrededor del 35% de los casos, y en 11%, el injerto fracasará completamente (que requieren la reanudación de la RRT y un posterior trasplante).[7]
Epidemiología
HSP ocurre con más frecuencia en niños que en adultos,[16] y generalmente sigue un infección del tracto respiratorio superior. La mitad de los pacientes afectados están por debajo de la edad de seis años, y el 90% en diez años. Se produce sobre dos veces más frecuente en los niños como las niñas.[5] El incidencia de HSP en niños es alrededor de 20 por 100.000 niños al año, por lo que es la vasculitis más frecuente en niños.[18]
Casos de HSP pueden ocurrir en cualquier momento durante todo el año, pero algunos estudios han encontrado que menos casos ocurren durante los meses de verano.[19]
Historia
La enfermedad es el nombre de Eduard Heinrich Henoch (1820-1910), un alemán pediatra (sobrino de Moritz Heinrich Romberg) y su profesor Johann Lukas Schönlein (1793-1864), quien lo describió en la década de 1860. Schönlein asociados la púrpura y la artritis y la púrpura de Henoch y la implicación gastrointestinal. Los ingleses médico William Heberden (1710-1801) y el dermatólogo Robert Willan (1757-1812) tenía ya la enfermedad descrita en 1802 y 1808, respectivamente, pero el nombre Enfermedad de Heberden – Willan ha caído en desuso. William Osler fue el primero en reconocer el subyacente alérgico mecanismo de HSP.[20]
Véase también
- Vasculitis de vasos pequeños cutánea
Referencias
- ^ a b c Grelo RP, Bolognia JL, Jorizzo JL (2007). Dermatología. St. Louis: Mosby. ISBN1-4160-2999-0.
- ^ a b c d Kraft DM, Mckee D, Scott C (1998). "La púrpura de Henoch-Schönlein: una revisión". Médico de familia americano 58 (2): 405 – 8, 411. PMID9713395.
- ^ a b c Saulsbury FT (1999). "Púrpura de Henoch-Schönlein en niños. Informe de 100 pacientes y revisión de la literatura". Medicine (Baltimore) 78 (6): 395-409. Doi:10.1097/00005792-199911000-00005. PMID10575422.
- ^ Fauci AS (1987). "269: los síndromes de Vasculitis". Braunwald E, Isselbacher KJ, Petersdorf RG, Wilson JD, Martin JB, Fauci AS. Libro de Harrison de la medicina interna 2 (11 Ed.). Colina de McGraw. p. 1441. ISBN0-07-079454-5.
- ^ a b c d e f g h i Saulsbury FT (2001). "Púrpura de Henoch-Schönlein". Current Opinion in Rheumatology 13 (1): 35 – 40. Doi:10.1097/00002281-200101000-00006. PMID11148713.
- ^ a b Shrestha S, Sumingan N, Tan J, et al (2006). "La púrpura de Henoch Schönlein con nefritis en adultos: indicadores pronósticos adversos en una población del Reino Unido". QJM 99 (4): 253-65. Doi:10.1093/qjmed/hcl034. PMID16565522.
- ^ a b c d e f g h i Rai A, C, Nast Adler S (01 de diciembre de 1999). "Nefritis de la púrpura de Henoch-Schönlein". Journal of the American Society of Nephrology 10 (12): 2637 – 44. PMID10589705.
- ^ Michel BA, Hunder GG, Bloch DA, Calabrese LH (1992). "Vasculitis de la hipersensibilidad y la púrpura de Henoch-Schönlein: una comparación entre los 2 trastornos". Revista de Reumatología 19 (5): 721-8. PMID1613701.
- ^ Mills JA, Michel BA, Bloch DA, et al (1990). "The American College of Rheumatology 1990 criterios para la clasificación de la púrpura de Henoch-Schönlein". Artritis y reumatismo 33 (8): 1114 – 21. Doi:10.1002/Art.1780330809. PMID2202310.
- ^ Colegio estadounidense de Reumatología. "criterios de 1990 para la clasificación de la púrpura de Henoch-Schönlein". de 2007-12-15.
- ^ Jennette JC, Falk RJ, Andrassy K, et al (1994). "Nomenclatura de vasculitides sistémicos. Propuesta de una conferencia de consenso internacional". Artritis y reumatismo 37 (2): 187 – 92. Doi:10.1002/Art.1780370206. PMID8129773.
- ^ Murali NS, George R, John GT, et al (2002). "Problemas de clasificación de la púrpura de Henoch Schonlein: una perspectiva indígena". Dermatolology clínica y Experimental 27 (4): 260-3. Doi:10.1046/j.1365-2230.2002.01063.x. PMID12139664.
- ^ Ozen S, Ruperto N, Dillon MJ, et al (julio de 2006). "EULAR/PReS respaldó" criterios de consenso * para la clasificación de infancia vasculitides. Anales de las enfermedades reumáticas 65 (7): 936 – 41. Doi:10.1136/ARD.2005.046300. PMC1798210. PMID16322081.
- ^ Weiss PF, Feinstein JA, Luan X, Burnham JM, Feudtner C (2007). "Efectos de corticosteroides en la púrpura de Henoch-Schönlein: una revisión sistemática". Pediatría 120 (5): 1079 – 87. Doi:10.1542/peds.2007-0667. PMC3525094. PMID17974746.
- ^ Chartapisak, W; Sauwalak, S; Hodson, EM; Willis, NS; Craig, JC (08 de julio de 2009). «Intervenciones para la prevención y tratamiento de enfermedades renales en la púrpura de Henoch-Schönlein (HSP).». Base de datos Cochrane de revisiones sistemáticas (en línea) (3): CD005128. Doi:10.1002/14651858.CD005128.pub2. PMID19588365.
- ^ a b Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García Fuentes M, González-Gay MA (1997). "Púrpura de Henoch-Schönlein en la adultez y la niñez: dos diferentes expresiones del mismo síndrome". Artritis y reumatismo 40 (5): 859 – 64. Doi:10.1002/Art.1780400513. PMID9153547.
- ^ Watson, L; Richardson, AR; Holt, RC; Jones, CA; Beresford, MW (enero de 2012). "Púrpura de Henoch Schoenlein--un 5 años revisar y propuso vía".. PLoS uno 7 (1): e29512. Doi:10.1371/Journal.pone.0029512. PMC3250434. PMID22235302. 15 de agosto de 2012.
- ^ Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR (2002). "Incidencia de la púrpura de Henoch-Schönlein, enfermedad de Kawasaki y vasculitides rara en los niños de diferentes orígenes étnicos". The Lancet 360 (9341): 1197 – 202. Doi:10.1016/S0140-6736 (02) 11279-7. PMID12401245.
- ^ Saulsbury FT (2002). "Epidemiología de la púrpura de Henoch-Schönlein". Diario de la Cleveland Clinic de medicina. 69 Suppl 2: SII87 – 9. PMID12086273.
- ^ Púrpura de Schönlein-Henoch en ¿Quién lo nombró?
| Wikimedia Commons tiene medios relacionados con Púrpura de Henoch-Schönlein. |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||