Electroforesis de proteínas

Electroforesis de proteínas es un método para el análisis de las proteínas en un fluido o de un extracto. La electroforesis se pueden realizar con un pequeño volumen de muestra en una serie de alternativas con o sin un soporte: Electroforesis en gel de poliacrilamida SDS (en Resumen: gel de electroforesis, página o electroforesis SDS), electroforesis de flujo libre, electroenfoque, isotachophoresis, electroforesis de afinidad, Inmunoelectroforesis en, counterelectrophoresis, y electroforesis capilar. Cada método tiene muchas variaciones individuales ventajas y limitaciones. Electroforesis en gel de se realiza a menudo en combinación con electroblotting immunoblotting dar información adicional sobre una proteína específica. Debido a limitaciones prácticas, electroforesis de proteínas generalmente no es la adecuada como método preparativo.
Contenido
- 1 Desnaturalización de métodos de gel
- 1.1 SDS-PAGE
- 2 Métodos nativos gel
- 2.1 Página nativa azul
- 2.2 Borrar Página nativa
- 2.3 Página continua nativa preparatoria cuantitativa
- 3 Sistemas pulmón
- 3.1 Electroforesis del gel del gradiente de SDS de proteínas
- 4 Visualización
- 5 Aplicaciones médicas
- 6 Véase también
- 7 Referencias
- 8 Enlaces externos
Desnaturalización de métodos de gel
SDS-PAGE
SDS-PAGE, dodecil sulfato de sodio electroforesis en gel de poliacrilamida, describe una colección de técnicas relacionadas para separar proteínas según su movilidad electroforética (una función de la longitud de una cadena polipeptídica y su carga) mientras que en el desnaturalizado Estado (desplegado). En la mayoría de las proteínas, la Unión del SDS a la cadena polipeptídica imparte una distribución uniforme de carga por unidad de masa, lo que resulta en un fraccionamiento por tamaño aproximado durante la electroforesis.
SDS es un agente detergente fuerte usado para desnaturalizar las proteínas nativas desplegado, individual polipéptidos. Cuando una mezcla de proteína se calienta a 100 ° C en presencia de SDS, la detergente se envuelve alrededor de la espina dorsal del polipéptido. En este proceso, las cargas intrínsecas de los polipéptidos se convierte en insignificante en comparación con las cargas negativas aportadas por SDS. Así los polipéptidos después del tratamiento se convierten en barra-como las estructuras que poseen una densidad de carga uniforme, que es la misma carga negativa neta por unidad de longitud. Las movilidades electroforéticas de las proteínas será una función lineal de la logaritmos de sus pesos moleculares.
Métodos nativos gel
Geles nativos, también conocida como no desnaturalizar los geles, analizan las proteínas que están todavía en su estado plegado. Así, la movilidad electroforética depende no sólo de la relación carga a masa, sino también a la forma física y tamaño de la proteína.
Página nativa azul
BN-PAGE es oriundo PÁGINA técnica, donde la Azul brillante de Coomassie colorante proporciona el necesario cargos a los complejos de proteína para la separación electroforética.[1][2] La desventaja de Coomassie es que en la Unión a las proteínas pueden actuar como un detergente causando complejos a disociar. Otro inconveniente es el potencial amortiguamiento de de quimioluminescencia (p. ej. en posteriores Western blot ensayos de detección o de actividad) o fluorescencia de proteínas con grupos prostéticos (p. ej. heme o clorofila) o con colorantes fluorescentes.
Borrar Página nativa
CN-PAGE (comúnmente denominado Página nativa) separa ácido soluble en agua y membrana proteínas en un poliacrilamida gel del gradiente. No utiliza ningún colorante cargado por lo que la movilidad electroforética de proteínas en la página de CN (en contraste con la técnica de cambio de carga BN-PAGE) se relaciona con la carga intrínseca de las proteínas.[3] La distancia de migración depende de la carga de la proteína, su tamaño y el tamaño del poro del gel. En muchos casos este método tiene menor resolución que página BN pero CN-PAGE ofrece ventajas cuando Coomassie tinte interferir con otras técnicas analíticas, por ejemplo se ha descrito como una técnica de separación de microescala muy eficiente para TRASTE Análisis.[4] También página de CN es más suave que BN-PAGE por lo que pueden mantener asambleas supramoleculares lábiles de proteína de la membrana complejos que son disociado en las condiciones de la página de la BN.
Página continua nativa preparatoria cuantitativa
En contraste con la página de CN y BN la doblado complejos de la proteína de interés separado limpio y predecible, ya que se mueven a través del gel de poliacrilamida tan rápidamente como las proteínas individuales, desnaturalizadas bajo no restrictivas condiciones. Las proteínas separadas continuamente se eluyeron en un eluyente fisiológica y transportadas a un colector de fracciones. En unas pocas fracciones de página específicos cofactores metálicos pueden identificarse y cuantificadas por técnicas de alta resolución. Las estructuras naturales de los aislados metaloproteinas es aclarada por la solución de NMR.[5]
Sistemas pulmón

Más separaciones de proteínas se realizaron mediante un "discontinuo" (o disco) tampón de sistema que mejora significativamente la nitidez de las bandas en el gel. Durante la electroforesis en un gel discontinuo sistema, un gradiente del ion se forma en la etapa temprana de la electroforesis que causa todas las proteínas para centrarse en una única banda sharp. La formación del gradiente iónico se logra seleccionando un valor de pH en el cual los iones del tampón de se cargan solamente moderado en comparación con las proteínas SDS-revestida. Estas condiciones proporcionan un ambiente en el que De Kohlrausch las reacciones determinan la conductividad molar. Como resultado, revestido de SDS proteínas se concentran a veces varias en una zona delgada del orden 19 μm en pocos minutos. En esta etapa todas las proteínas migran a la misma velocidad de migración por isotachophoresis. Esto ocurre en una región del gel que tiene poros más grandes de modo que la matriz de gel no retarda la migración durante el evento de enfoque o "apilamiento".[6][7] Separación de las proteínas por tamaño se consigue en la parte inferior, "resolver" región del gel. El gel de resolución por lo general tiene un tamaño de poro mucho más pequeño, que conduce a un efecto de tamizado que ahora determina la movilidad electroforética de las proteínas. Al mismo tiempo, la parte de separación del gel también tiene un valor de pH en el cual los iones del tampón en promedio transportar una carga mayor, haciendo que "escapar" de las proteínas de cubierta de SDS y eliminar el gradiente de iones y el apilamiento efecto de tal modo.
Un sistema de tampón discontinuo muy extendido es el tris glicina o "Laemmli"sistema que acumula en un pH de 6.8 y resuelve en un pH de ~8.3-9.0. Un inconveniente de este sistema es que pueden promover estos valores de pH disulfuro de formación de enlace entre cisteína residuos en las proteínas porque la pKa de rangos de cisteína de 8-9 y porque el agente reductor presente en el tampón de carga no migra conjuntamente con las proteínas. Los avances recientes en tecnología de almacenamiento en búfer aliviar este problema resolviendo las proteínas a un pH muy por debajo del pKa de cisteína (p. ej., bis-trispH 6,5) y agentes de reducción (e.g. bisulfito de sodio) que se mueven en el gel por delante de las proteínas para mantener un ambiente reductor. Un beneficio adicional del uso de tampones con valores de pH más bajos es que el gel de acrilamida es más estable a valores de pH más bajos, por lo que los geles pueden almacenarse durante largos periodos de tiempo antes de su uso.[8][9]
Electroforesis del gel del gradiente de SDS de proteínas
Como se aplica el voltaje, los aniones (y las moléculas negativamente cargadas de la muestra) migran hacia el electrodo positivo (ánodo) en la cámara baja, es el principal ión Cl− (alta movilidad y elevada concentración); glicinato de sodio es el ion que se arrastra (baja movilidad y concentración baja). SDS-proteína partículas no migran libremente en la frontera entre el Cl− del buffer gel y el Gly− del buffer catódico. Friedrich Kohlrausch encontró que Ley de Ohm también se aplica a disuelto electrólitos. Debido a la caída de tensión entre el Cl− y glicina-buffers, proteínas están comprimidos (apilados) en capas delgadas de micrómetro.[10] El límite se mueve a través de un gradiente de poro y la pila de la proteína gradualmente se dispersa debido a un aumento de la resistencia friccional de la matriz de gel. Apilado y desapilado se produce continuamente en el gel de gradiente para cada proteína en una posición diferente. Para una proteína completa desapilado de la concentración del gel de poliacrilamida debe exceder 16% T. El sistema de dos geles de "Laemmli" es un simple gel del gradiente. La discontinuidad del pH de los amortiguadores es de ninguna importancia para la calidad de separación, y no es necesario un "apilado-gel" con un pH diferente.
Visualización
La mancha de proteína más popular es Azul brillante de Coomassie. Es un colorante aniónico, que se une no específicamente a las proteínas. Las proteínas en el gel son fijas por ácido acético y simultáneamente manchadas. El exceso de tinte incorporado en el gel puede eliminarse por extracción con la misma solución sin el tinte. Las proteínas se detectan como bandas azules sobre un fondo claro.
Cuando el método más sensible que la tinción de Coomassie es necesario normalmente se utiliza la tinción de plata. Tinción de plata es un procedimiento sensible para detectar pequeñas cantidades de proteínas en geles, pero también puede visualizar ácidos nucleicos o los polisacáridos.
Semejantemente como en ácido nucleico gel de electroforesis, seguimiento de tinte es de uso frecuente. Colorantes aniónicos de una movilidad electroforética conocida generalmente están incluidos en el tampón de muestra. Un tinte muy común de seguimiento es Azul de bromofenol. Este colorante es de color en el álcali y de pH neutro y se carga negativamente una pequeña molécula que se mueve hacia el ánodo. Ser una molécula altamente móvil que se mueve por delante de la mayoría de las proteínas.
Aplicaciones médicas

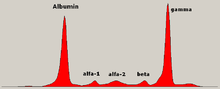
En medicina, electroforesis de proteínas es un método de análisis de la proteínas principalmente en suero de la sangre (plasma de sangre no es conveniente[citación necesitada]). Antes del uso generalizado de electroforesis en gel de, electroforesis de proteínas se realizó electroforesis de flujo continuo (en papel) o como inmunoelectroforesis.
Tradicionalmente, dos clases de proteínas de la sangre se consideran: albúmina de suero y globulina. Son generalmente iguales en proporción, pero albúmina como una molécula es mucho más pequeña y ligera, negativamente cargada, llevando a una acumulación de albúmina en el gel de electroforesis. Un pequeño grupo antes de albúmina representa transtiretina (también llamada prealbúmina). Algunas formas de medicación o cuerpo productos químicos pueden causar su propia banda, pero generalmente es pequeña. Se observan bandas anormales (picos) en gammapatía monoclonal de significación indeterminada y mieloma múltipley son útiles en el diagnóstico de estas condiciones.
Las globulinas se clasifican por su patrón de bandas (con sus principales representantes):
- El alfa (Α) banda consta de dos partes, 1 y 2:
- α1 - α1-antitripsina, Α1-glicoproteína ácida.
- α2 - haptoglobina, α2-macroglobulina, α2-antiplasmina, ceruloplasmina.
- El Beta (Β) banda- transferrina, LDL, complemento
- El gamma (Γ) banda- inmunoglobulina (IgA, IgD, IgE, IgG e IgM). Paraproteínas (en el mieloma múltiple) suelen aparecer en esta banda.
Normal presente procedimiento médico implica determinación de numerosas proteínas en plasma incluyendo las hormonas y las enzimas, algunas de ellas también determinado por electroforesis. Sin embargo, electroforesis en gel es principalmente una herramienta de investigación, también cuando el sujeto es proteínas de la sangre.
Véase también
- Electroforesis de afinidad
- Electroblotting
- Electroenfoque
- Electroforesis en gel de poliacrilamida, página o electroforesis en gel de
- Inmunoelectroforesis en
- Inmunofijación
- Electroforesis nativa
- QPNC-PAGE
- Paraproteína
- Proteólisis rápida paralela (FASTpp)[11]
Referencias
- ^ Schägger, H.; Jagow, G. (1991). «Electroforesis nativa azul para el aislamiento de complejos de proteínas de membrana en forma enzimáticamente activa». Anal. Biochem. 199 (2): 223-231. doi:10.1016/0003-2697 (91) 90094-A. PMID1812789
- ^ Wittig, I.; Braun, H.P.; Schägger, H. (2006). «Página nativa azul». Protoc nacional. 1 (1): 418-428. doi:10.1038/nprot.2006.62. PMID17406264
- ^ Wittig, I.; Schägger, H. (Nov de 2005). "Ventajas y limitaciones de la página claro nativo". Proteómica 5 (17): 4338 – 46. doi:10.1002/PMIC.200500081. PMID16220535.
- ^ P.D. de Gavin, R.J. Devenish, Prescott M. (2003). "Traste revela cambios en la F1– interacción de tallo del estator durante la actividad de F1F0-ATP sintasa ". Acta de Biochim Biophys 1607 (2-3): 167-79. doi:10.1016/j.bbabio.2003.09.013.
- ^ Kastenholz, B. (2006). "Contribuciones importantes de un nuevo procedimiento de electroforesis (QPNC-PAGE) cuantitativa preparativa nativos continua gel de poliacrilamida para elucidar metal cofactor metabolismos en enfermedades mal plegamiento de la proteína – una teoría". Proteína Pept Lett 13 (5): 503-8. doi:10.2174/092986606776819637. PMID16800806.
- ^ L de Ornstein (diciembre de 1964). "Discos electroforesis. I. antecedentes y teoría". Anales de la Academia de Ciencias de Nueva York 121 (2): 321-349. doi:10.1111/j.1749-6632.1964.tb14207.x. PMID14240533.
- ^ Davis BJ (diciembre de 1964). "Discos electroforesis. 2, método y aplicación a las proteínas del suero humano". Ann. Nueva York Acad. Sci 121 (2): 404 – 427. doi:10.1111/j.1749-6632.1964.tb14213.x. PMID14240539.
- ^ Schägger H, von Jagow G (1987). «Tricina-sodio dodecil sulfato-electroforesis gel de poliacrilamida para la separación de proteínas en el rango de 1 a 100 kDa». Anal Biochem. 166 (2): 368-379. doi:10.1016/0003-2697 (87) 90587-2. PMID2449095.
- ^ Wiltfang J, Arold N, V Neuhoff (1991). "Un nuevo sistema de amortiguación multifásica para sodio dodecil sulfato-poliacrilamida gel electroforesis de proteínas y péptidos con masas moleculares 100.000-1000 y su detección con sensibilidad picomolar." Electroforesis 12 (5): 352-366. doi:10.1002/ELPS.1150120507. PMID1718736.
- ^ F de Kohlrausch (1897). "Ueber concentraciones-Verschiebungen durch Electrolyse im Inneren von Lösungen und Lösungsgemischen". Ann.J.Phys.u.Chem. 62 (10): 209-239. doi:10.1002/andp.18972981002.
- ^ Minde DP (2012). "Determinación de estabilidad biofísica de proteínas en lisados por un ensayo de proteólisis rápida, FASTpp". PLOS ONE 7 (10): e46147. doi:10.1371/journal.pone.0046147. PMC3463568. PMID23056252.
Enlaces externos
- El texto completo había editado por Niels H. Axelsen en Revista escandinava de Inmunología, volumen 1975 suplemento 4. Este es el texto de la opción para inmunoelectroforesis.
- Electroforesis del gel de proteína nativa discontinua
- Instalación de purificación de proteínas
- Recurso educativo para electroforesis de proteínas
- Electroforesis de proteínas
|
||||||||||||||||||||||
Otras Páginas
- Lista de casos del Tribunal Supremo de Reino Unido
- IPhone 5
- Samsung Galaxy Y
- WDV
- Distrito escolar independiente de Cedar Hill
- Lista de unidades estratigraficas fosiliferas en Nunavut
- Deportes Juegos de mundo
- Vinagre
- Rat Race (pelicula)
- Cuerpo del glomus
- Pedro Zamora (articulos de la categoria con hCards)