Electroforesis en gel de

Aparato de electroforesis gel, un gel de agarosa se coloca en este cuadro lleno de tampón y se aplica un campo eléctrico a través de la alimentación en la parte trasera. El polo negativo está en el otro extremo (cable negro), por lo que el ADN migra hacia el ánodo (alambre rojo).
|
|
| Clasificación | Electroforesis |
|---|---|
| Otras técnicas de | |
| Relacionados con la | Electroforesis capilar SDS-PAGE Electroforesis en gel bidimensional Electroforesis de gel gradiente de temperatura |
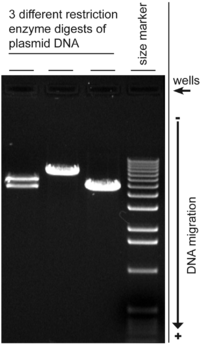
Electroforesis en gel de es un método de separación y análisis de macromoléculas (DNA, RNA y proteínas) y sus fragmentos, basados en su tamaño y carga. Se utiliza en química clínica para separar las proteínas por carga o tamaño (agarosa IEF, esencialmente independiente del tamaño) y en Bioquímica y biología molecular para separar una población mixta de DNA y RNA fragmentos de longitud, para estimar el tamaño de DNA y RNA fragmentos o separar proteínas por carga.[1]
Moléculas de ácido nucleico se separan mediante la aplicación de un campo eléctrico para mover las moléculas negativamente cargadas a través de una matriz de agarosa u otras sustancias. Las moléculas más cortas se mueven más rápido y migran más allá de los más largos porque las moléculas más cortas migran más fácilmente a través de los poros del gel. Este fenómeno se llama tamizado.[2]
Las proteínas se separan por cargo en agarosa porque los poros del gel están demasiado grandes para tamizar las proteínas.
Electroforesis en gel puede utilizarse también para la separación de las nanopartículas.
Electroforesis en gel utiliza un gel como un medio de anticonvective o tamizado medio durante la electroforesis, el movimiento de una partícula cargada en un campo eléctrico. Geles suprimen la convección térmica causada por la aplicación del campo eléctrico y también pueden actuar como un medio de tamizado, retardando el paso de moléculas; geles también simplemente pueden servir para mantener la separación final, por lo que puede aplicarse un tinte de electroforesis de post.[3] Electroforesis del Gel de DNA se realizan generalmente con fines analíticos, a menudo después de la amplificación de ADN mediante la POLIMERIZACIÓN EN CADENA, pero puede utilizarse como un previa preparación técnica para uso de otros métodos tales como espectrometría de masas, RFLP, POLIMERIZACIÓN EN CADENA, clonación, Secuencia de la DNA, o Borrar meridional para la caracterización de más.
Contenido
- 1 Base física
- 2 Tipos de gel
- 2.1 Agarosa
- 2.2 Poliacrilamida
- 2.3 Almidón
- 3 Condiciones de gel
- 3.1 La desnaturalización
- 3.2 Nativo
- 4 Almacenadores intermediarios
- 5 Visualización
- 6 Procesamiento descendente
- 7 Aplicaciones
- 7.1 Ácidos nucleicos
- 7.2 Proteínas
- 8 Historia
- 9 Véase también
- 10 Referencias
- 11 Enlaces externos
Base física
En términos simples, electroforesis es un proceso que permite la clasificación de moléculas según el tamaño. Usando un campo eléctrico, las moléculas (por ejemplo, ADN) se pueden hacer para moverse a través de un gel de Agar o poliacrilamida. El campo eléctrico consiste en una carga negativa en un extremo que empuja a las moléculas a través del gel y una carga positiva en el otro extremo que atrae las moléculas a través del gel. Las moléculas se ordenan son dispensadas en un pozo en el material de gel. El gel se coloca en una cámara de electroforesis, que se conecta a una fuente de energía. Cuando se aplica la corriente eléctrica, moverse más lentamente las moléculas más grandes a través del gel mientras que las moléculas pequeñas se mueven más rápido. Las moléculas de tamaños diferentes forman distintas bandas en el gel.[citación necesitada]
El término"gel"en este caso se refiere a la matriz utilizada para contener y luego separar las moléculas objetivo. En la mayoría de los casos, el gel es un polímero reticulado cuya composición y porosidad se elige basado en el peso específico y la composición del objetivo a analizar. Al separar proteínas o pequeño ácidos nucleicos (DNA, RNA, o oligonucleótidos) el gel se compone generalmente de diferentes concentraciones de acrilamida y un vinculante, produciendo diferentes tamaño de malla de redes de poliacrilamida. Al separar los ácidos nucleicos más grandes (más de unos pocos cientos bases), la matriz preferida es purificada agarosa. En ambos casos, el gel forma una matriz sólida y porosa. Acrilamida, en contraste con poliacrilamida, es un neurotoxina y deben ser manejados utilizando precauciones de seguridad apropiadas para evitar el envenenamiento. Agarosa se compone de cadenas no ramificadas largo de carbohidratos sin cargar sin Cruz enlaces resultando en un gel con poros grandes que permiten la separación de macromoléculas y complejos macromoleculares.[citación necesitada]
"Electroforesis"se refiere a la fuerza electromotriz (EMF) que se utiliza para mover las moléculas a través de la matriz de gel. Colocando las moléculas en los pocillos en el gel y aplicar un campo eléctrico, las moléculas se mueven a través de la matriz a diferentes tasas, determinadas en gran medida por su masa cuando la carga a la relación entre la masa (Z) de todas las especies es uniforme. Sin embargo cuando los cargos no son uniforme todos entonces, el campo eléctrico generado por el procedimiento de electroforesis afectará a las especies que tienen diferentes cargas y por lo tanto, atraerá a las especies según sus cargos siendo lo contrario. Especies que están cargadas positivamente (cationes) migrarán hacia el cátodo que se carga negativamente. Si las especies están cargadas negativamente (aniones) migran hacia el ánodo cargado positivamente.[4]
Si varias muestras han sido cargadas en pozos adyacentes en el gel, corren paralelas en carriles individuales. Dependiendo del número de moléculas diferentes, cada carril muestra separación de los componentes de la mezcla original como uno o más grupos distintos, una banda por componente. Separación incompleta de los componentes puede conducir a la superposición de bandas o frotis indistinguibles representando múltiples componentes sin resolver.[citación necesitada] Bandas en diferentes carriles que terminan a la misma distancia desde la parte superior contienen moléculas que pasan a través del gel con la misma velocidad, que generalmente significa que son aproximadamente del mismo tamaño. Hay marcadores de peso molecular tamaño disponibles contienen una mezcla de moléculas de tamaños conocidos. Si tal marcador fue ejecutado en un carril del gel paralelo a las muestras desconocidas, las bandas observadas pueden compararse a los de lo desconocido para determinar su tamaño. La distancia que viaja una banda aproximadamente es inversamente proporcional al logaritmo del tamaño de la molécula.[citación necesitada]
Hay límites a las técnicas electroforéticas. Desde pasar actual a través de un gel causa calentamiento, geles pueden derretirse durante la electroforesis. Electroforesis se realizan en soluciones tampón a reducir los cambios de pH debido al campo eléctrico, que es importante porque la carga de DNA y RNA depende de pH, pero durante demasiado tiempo puede agotar la capacidad tampón de la solución. Además, diferentes preparaciones de material genético no pueden migrar constantemente con los demás, por razones morfológicas o de otros.
Tipos de gel
Los tipos de gel que se utiliza más típicamente son geles de poliacrilamida y agarosa. Cada tipo de gel es adecuado para diferentes tipos y tamaños de analito. Geles de poliacrilamida se utilizan generalmente para las proteínas y tienen muy alta energía de resolución para fragmentos pequeños de DNA (5-500 PB). Geles de agarosa por otro lado tienen menor poder de resolución para el ADN, pero tienen mayor rango de separación y por lo tanto se utilizan para fragmentos de ADN de generalmente 50-20.000 bp en tamaño, pero es posible con resolución de más de 6 Mb electroforesis en gel de campo pulsátil (PFGE).[5] Geles de poliacrilamida se ejecutan en una configuración vertical, mientras que geles de agarosa son típicamente horizontalmente en un modo de submarino. También difieren en su metodología del casting, como agarosa térmicamente, mientras que formas de poliacrilamida en una reacción química de polimerización.
Agarosa
Agarosa los geles se hacen de lo natural polisacárido polímeros extraído de algas marinas. Agarosa los geles son fácilmente del molde y maneja en comparación con otras matrices, ya que el gel es un cambio físico más que químico. Las muestras se recuperan fácilmente. Una vez terminado el experimento, el gel resultante puede guardarse en una bolsa de plástico en el refrigerador.
Geles de agarosa no tienen un tamaño de poro uniforme, pero son óptimos para la electroforesis de las proteínas que son más grandes que 200 kDa.[6] Electroforesis en gel de agarosa pueden utilizarse también para la separación de fragmentos de ADN que van desde 50 base par a varias megabases (millones de bases), el mayor de los cuales necesita aparatos especializados. La distancia entre las bandas de ADN de diferentes longitudes es influenciada por el porcentaje agarosa en el gel, con porcentajes más altos que requieren largo plazo de las épocas, a veces días. En cambio, geles de agarosa alto porcentaje deben realizarse con un electroforesis en campo pulsado (PFE), o electroforesis de la inversión del campo.
"La mayoría de geles de agarosa se hacen con entre 2% y el 0,7% (buena separación o resolución de fragmentos de ADN de grandes 5 – 10kb) (resolución buena para pequeños 0.2 – 1 kb fragmentos) agarosa disuelta en buffer de electroforesis. Hasta el 3% puede ser utilizado para separar fragmentos muy pequeños pero vertical gel de poliacrilamida en este caso es más apropiado. Geles de bajo porcentaje son muy débiles y pueden romperse al intentar levantarlas. Alto porcentaje los geles son a menudo frágiles pero no uniformemente. 1% geles son comunes para muchas aplicaciones".[7]
Poliacrilamida
Poliacrilamida gel de electroforesis (PAGE) se utilizan para separar proteínas que varían en tamaño desde 5 hasta 2.000 kDa debido al tamaño de poro uniforme proporcionado por el gel de poliacrilamida. Tamaño de los poros se controla modulando las concentraciones de acrilamida y bis-acrilamida en polvo utilizado en la creación de un gel. Debe tenerse cuidado al crear este tipo de gel, como la acrilamida es una neurotoxina potente en sus formas líquidas y en polvo.
Tradicional Secuencia de la DNA técnicas tales como MAXAM-Gilbert o Sanger métodos utilizan geles de poliacrilamida para separar fragmentos de ADN que difieren por un solo par de base de longitud por lo que se pudo leer la secuencia. Métodos de separación de ADN más modernos ahora utilizan geles de agarosa, excepción de ADN particularmente pequeño fragmentos. Actualmente se utiliza más a menudo en el campo de la Inmunología y análisis de proteínas, a menudo se utiliza para separar proteínas diferentes o isoformas de la misma proteína en diferentes bandas. Estos pueden ser transferidos a un nitrocelulosa o PVDF membrana para ser sondeado con anticuerpos y marcadores correspondientes, como en un Western blot.
Por lo general resolución de los geles se fabrican en 6%, 8%, 10%, 12% o 15%. Stacking gel (5%) se vierte sobre el gel de resolución y se inserta un peine de gel (que forma los pozos y define los carriles donde se colocarán las proteínas, tampón y escaleras). El porcentaje elegido depende del tamaño de la proteína que se desea identificar o sonda en la muestra. Cuanto menor sea el peso conocido, cuanto mayor sea el porcentaje que se debe utilizar. Cambios en el sistema buffer del gel pueden ayudar a resolver más proteínas de tamaño muy pequeño.[8]
Almidón
Parcialmente hidrolizado de almidón de patata se hace por otro medio no tóxico para electroforesis de proteínas. Los geles son un poco más opacos que la acrilamida o agarosa. Las proteínas desnaturalizadas no se pueden separar según tamaño y carga. Ellos son visualizadas mediante tinción Napthal Black o Amido Black. Concentraciones de gel de almidón típicos son 5% a 10%.[9][10][11]
Condiciones de gel
La desnaturalización
La desnaturalización geles se ejecutan bajo las condiciones que alteran la estructura natural del analito, causando que se despliegan en una cadena lineal. Así, la movilidad de cada uno macromolécula depende solamente de su longitud y su relación masa / carga. Así, los niveles secundarios, terciarios y cuaternarios de estructura de biomoléculas se interrumpe, dejando sólo la estructura primaria a analizar.
Los ácidos nucleicos son desnaturalizados a menudo incluyendo urea en el buffer, mientras que las proteínas son desnaturalizadas mediante dodecil sulfato de sodio, generalmente como parte de la SDS-PAGE proceso. Para la completa desnaturalización de las proteínas, también es necesario reducir el covalente enlaces de disulfuro que estabilice su terciario y estructura cuaternaria, llama a un método reducción de la página. Condiciones reductoras son mantenidas generalmente por la adición de beta-mercaptoetanol o Ditiotreitol. Para el análisis general de las muestras de proteína, reducción de la página es la forma más común de electroforesis de proteínas.
Condiciones de desnaturalización son necesarias para la adecuada estimación del peso molecular de RNA. ARN es capaz de formar más interacciones intramoleculares de ADN que puede resultar en cambio de su movilidad electroforética. Urea, DMSO y glioxal lo más a menudo utilizan agentes desnaturalizantes para interrumpir la estructura del RNA. Originalmente, altamente tóxico metilmercurio hidróxido era de uso frecuente en electroforesis de RNA, la desnaturalización[12] pero puede ser método de elección para algunas muestras.[13]
Desnaturalizando electroforesis del gel se utiliza en la DNA y el RNA métodos basados en el patrón de bandas DGGE (electroforesis de gel gradiente desnaturalización),[14] TGGE (electroforesis de gel gradiente de temperatura) y TTGE (electroforesis en gradiente de temperatura temporal).[15]
Nativo
Geles nativos se ejecutan en condiciones no desnaturalizantes, para que se mantenga la estructura del analito. Esto permite que el tamaño físico de la doblada o montar complejo afectan la movilidad, permitiendo para el análisis de los cuatro niveles de la estructura biomolecular. Para muestras biológicas, detergentes se utilizan en la medida en que son necesarias para Lyse membranas lipídicas En célula. Siguen siendo complejos — en su mayor parte — asociados y doblado como serían en la célula. Sin embargo, una desventaja, es que no pueden separar complejos limpiamente o como era de esperarse, ya que es difícil predecir cómo la forma y el tamaño de la molécula afectará su movilidad.
A diferencia de los métodos de desnaturalización, electroforesis nativa no utilizan un la desnaturalización agente. Las moléculas se separen (generalmente proteínas o ácidos nucleicos) por lo tanto difieren no sólo en masa molecular y carga intrínseca, pero también el área transversal y así experiencia diferentes fuerzas electroforética depende de la forma de la estructura global. Para las proteínas, ya que permanecen en el estado nativo puede visualizar por proteína general reactivos de tinción, pero también por la coloración de enzima-ligado específico.
Nativo electroforesis en gel se utiliza normalmente en proteómica y Metalómica. Sin embargo, PAGE nativa también se utiliza para analizar genes (ADN) para mutaciones desconocidas como en Polimorfismo de la conformación del solo filamento.
Almacenadores intermediarios
Tampones de electroforesis en gel se utilizan para proporcionar iones que transportan una corriente y para mantener el pH en un valor relativamente constante. Hay un número de buffers utilizados para electroforesis. El ser más común, para los ácidos nucleicos Acetato/Tris/EDTA (TAE), Borato/Tris/EDTA (TBE). Se han propuesto muchos otros buffers, por ejemplo borato de litio, que casi nunca se utiliza, basado en las citas de Pubmed (LB), iso eléctrica histidina, pK emparejado almacenadores intermediarios de la mercancía, etc..; en la mayoría de los casos la justificación pretendida corriente baja (menos calor) y o emparejado movilidades de iones, que conduce a vida más larga de búfer. Borato es problemático; Borato puede polimerizarse o interactuar con dioles cis como los que se encuentran en el ARN. TAE tiene la capacidad de amortiguación más baja pero proporciona la mejor resolución para el ADN más grande. Esto significa una tensión más baja y más tiempo, pero un mejor producto. LB es relativamente nueva y es ineficaz en la resolución de fragmentos mayores de 5 kbp; Sin embargo, con su baja conductividad, una tensión mucho más alta podría utilizarse (hasta 35 V/cm), que significa un menor tiempo de análisis por electroforesis rutinaria. Tan bajos como una diferencia de tamaño de base par podría resolverse en gel de agarosa al 3% con un medio de conductividad extremadamente baja (borato de litio 1 mM).[17]
Más separaciones de proteínas SDS-PAGE se realizan utilizando un "discontinuo" (o disco) sistema del almacenador intermediario mejora significativamente la nitidez de las bandas en el gel. Durante la electroforesis en un gel discontinuo sistema, se forma un gradiente de iones en la etapa temprana de la electroforesis que causa todas las proteínas para centrarse en una única banda sharp en un proceso llamado isotachophoresis. Separación de las proteínas por tamaño se consigue en la parte inferior, "resolver" región del gel. El gel de resolución por lo general tiene un tamaño de poro mucho más pequeño, que conduce a un efecto de tamizado que ahora determina la movilidad electroforética de las proteínas.
Visualización
Una vez finalizada la electroforesis, las moléculas en el gel pueden ser manchado para hacerlos visibles. ADN puede visualizarse utilizando bromuro de etidio que, cuando se intercalan en el ADN, es fluorescente bajo radiación ultravioleta luz, mientras que la proteína se puede visualizar usando tinción de plata o Azul brillante de Coomassie colorante. También podrán utilizarse otros métodos para visualizar la separación de los componentes de la mezcla en el gel. Si contienen las moléculas a separar radioactividad, por ejemplo, en un Secuencia de la DNA gel, un autoradiogram se puede grabar del gel. Fotografías puede tomarse de los geles, a menudo utilizando un Gel Doc sistema.
Procesamiento descendente
Después de la separación, un método de separación adicional puede utilizarse, tales como isoeléctrica o SDS-PAGE. El gel será físicamente cortado y los complejos de la proteína extraídos de cada parte por separado. Cada extracto luego puede ser analizadas, tales como por péptido masa fingerprinting o secuenciación de péptidos de novo después de la digestión en gel. Esto puede proporcionar una gran cantidad de información sobre la identidad de las proteínas en un complejo.
Aplicaciones

- Estimación del tamaño de las moléculas de ADN después de digestión de la enzima de restricción, por ejemplo, en mapeo de restricción de ADN clonado.
- Análisis de la POLIMERIZACIÓN EN CADENA productos, por ejemplo en molecular diagnóstico genético o huella dactilar genética
- Separación de la DNA genomic restringida antes Transferencia de Southern, o de ARN antes de Transferencia Norte.
Electroforesis en gel se utiliza en medicina forense, biología molecular, genética, Microbiología y Bioquímica. Los resultados pueden ser analizados cuantitativamente por visualizar el gel con luz UV y un dispositivo de proyección de imagen del gel. La imagen se graba con una cámara de computadora operado, y la intensidad de la banda o punto de interés es medido y se compara contra el estándar o marcadores cargados en el mismo gel. La medición y análisis se realizan en su mayoría con software especializado.
Dependiendo del tipo de análisis se realiza, otras técnicas se aplican a menudo en conjunción con los resultados de la electroforesis en gel, proporcionando una amplia gama de aplicaciones específicas de campo.
Ácidos nucleicos
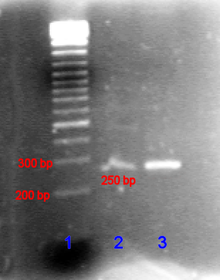
En el caso de los ácidos nucleicos, la dirección de migración, de negativo a los electrodos positivos, es debido a la carga negativa natural por su azúcar-fosfato de columna vertebral.[18]
Fragmentos de ADN de doble hebra se comportan naturalmente como varillas largas, así su migración a través del gel es en relación a su tamaño o por fragmentos cíclicos, su radio de giro. ADN circular como plásmidos, sin embargo, puede mostrar bandas múltiples, la velocidad de migración dependerá si es relajado o superenrollado. ADN o ARN monocatenario tienden a doblar para arriba en moléculas con formas complejas y migran por el gel de manera complicada basada en su estructura terciaria. Por lo tanto, los agentes que interrumpen la enlaces de hidrógeno, tales como hidróxido de sodio o formamida, se utiliza para desnaturalizar los ácidos nucleicos y hacer que se comportan como barras largas otra vez.[19]
Electroforesis en gel de gran DNA o RNA generalmente se realiza electroforesis en gel de agarosa. Ver el "Método de terminación de cadena"página para obtener un ejemplo de un poliacrilamida Gel de secuenciación de ADN. Caracterización a través de la interacción ligando de ácidos nucleicos o fragmentos puede realizarse por turno de movilidad electroforesis de afinidad.
Electroforesis de las muestras de RNA pueden utilizarse para comprobar la contaminación del ADN genómica y degradación del RNA. ARN de los organismos eucariotas muestra distintas bandas de 28s y 18s rRNA, el 28s banda siendo aproximadamente dos veces tan intenso como el 18s banda. Degrada el RNA ha menos agudamente definido bandas, tiene un aspecto sucio y cociente de la intensidad es menor que 2:1.
Proteínas
Proteínas, a diferencia de los ácidos nucleicos, pueden tener cargas diferentes y formas complejas, por lo tanto puede no migran en el gel de poliacrilamida en tasas similares, o en todos, al colocar un negativo a los CEM positivo en la muestra. Las proteínas por lo tanto, son generalmente desnaturalizado en presencia de un detergente tales como dodecil sulfato de sodio (SDS) que recubre las proteínas con carga negativa.[3] Generalmente, la cantidad de SDS obligado es relativo al tamaño de la proteína (generalmente 1,4 g SDS por gramo de proteína), por lo que la resultante desnaturalizado proteínas tienen una carga global negativa, y todas las proteínas tienen una carga similar a la relación entre la masa. Desnaturalizado las proteínas actuar como varillas largas en lugar de tener una forma compleja de la terciaria, la tasa en la que el SDS resultante cubrió proteínas migran en el gel es relativo solamente a su tamaño y su carga ni forma.[3]
Proteínas generalmente se analizan por sodio dodecil sulfato poliacrilamida gel electroforesis (SDS-PAGE), por electroforesis nativa, por () electroforesis cuantitativa preparativa nativos continua gel de poliacrilamidaQPNC-PAGE), o por Electroforesis en 2-D.
Caracterización a través de la interacción ligando puede ser realizado por electroblotting o por electroforesis de afinidad en agarosa o por electroforesis capilar en cuanto a la estimación de constantes de enlace y determinación de características estructurales como glycan contenido a través de lectina enlace.
Historia
- década de 1930, los primeros informes del uso de sacarosa para electroforesis en gel de
- 1955 – la introducción de almidón geles, separación de mediocre
- 1959 – la introducción de acrilamida geles; discos electroforesis (Ornstein y Davis); control preciso de los parámetros de los poros tales como tamaño y estabilidad; y (Raymond y Weintraub)
- 1966 – Agar geles de[20]
- 1969 – introducción de la desnaturalización agentes especialmente SDS separación de proteína subunidad (Weber y Osborn)[21]
- 1970 – Laemmli separados 28 componentes de Fago T4 usando un gel de apilamiento y SDS
- 1972 – agarosa geles con tinción de bromuro de etidio[22]
- 1975 – 2-dimensional geles (O'Farrell); isoeléctrica electroforesis en gel de SDS entonces
- 1977 – secuencia geles de
- 1983 – electroforesis en gel de campo pulsátil permite la separación de moléculas grandes de ADN
- 1983 – introducción de electroforesis capilar
- 2004 – estándar tiempo de la polimerización de geles de acrilamida permite la separación limpia y predecible de proteínas nativas[23]
Un libro de 1959 en la electroforesis por Milán Bier cita referencias desde el siglo XIX.[24] Sin embargo, Herrerías de Oliver hecho importantes contribuciones. Estados de Bier: "el método de fraguas... encuentra gran aplicación debido a su singular poder separatory." Tomado en contexto, Bier implica claramente que el método de Fraguas es una mejora.
Véase también
- Historia de la electroforesis
- Ensayo de cambio de movilidad electroforética
- Extracción del gel
- Isoeléctrica
- Electroforesis en gel de campo pulsátil
- Electroforesis en gel bidimensional
- QPNC-PAGE
- SDS-PAGE
- SDD-EDAD
- Mancha blanca /negra norteña
- Western blot
- Mancha Oriental
- Zymography
- Electroforesis de afinidad
- Proteólisis rápida paralela (FASTpp)
[25]
Referencias
- ^ Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV (2003). "Agregados de priones de la levadura [PSI +] están formadas por polímeros de Sup35 pequeños fragmentados por Hsp104." Diario de química biológica 278 (49): 49636 – 43. doi:10.1074/jbc. M307996200. PMID14507919.
- ^ Sambrook J, Russel DW (2001). Clonación molecular: Un Manual de laboratorio 3ª Ed. fría primavera puerto prensa de laboratorio. Cold Spring Harbor, NY.
- ^ a b c Berg JM, Tymoczko JL Stryer L (2002). Bioquímica (5ª ed.). Freeman de WH. ISBN0-7167-4955-6.
- ^ Robyt, John F. & blanco, Bernard J. (1990). Práctica y la teoría de técnicas bioquímicas. Prensa de Waveland. ISBN0-88133-556-8.
- ^ Tom Maniatis, E. F. Fritsch y José Sambrook. "Capítulo 5, protocolo 1". Clonación molecular - un Manual de laboratorio 1 (3ª Ed.). p. 5.2-5.3. ISBN978-0879691363.
- ^ Smisek, L. D.; Hoagland, D. A. (1989). "Gel de agarosa electroforesis de alto peso molecular, polielectrolitos sintéticos". Macromoléculas 22 (5): 2270. doi:10.1021/ma00195a048.
- ^ "Electroforesis en gel de agarosa (método básico)". Protocolos biológicos. 23 de agosto 2011.
- ^ Schägger, Hermann (2006). "Tricina-SDS-PAGE". Protocolos de la naturaleza 1 (1): 16 – 22. doi:10.1038/nprot.2006.4. PMID17406207.
- ^ Gordon, A.H. (1975). Electroforesis de proteínas en geles de poliacrilamida y almidón. Nueva York: Americano Elsevier Publishing Company, Inc.
- ^ Herrerías, o. (1955). "Zona de electroforesis en geles de almidón: las variaciones en las proteínas de suero de adultos normales del grupo". Biochem. J. 61 (4): 629-641. PMC1215845. PMID13276348.
- ^ Wraxall, B.G.D.; Culliford, B.J. (1968). "Un capa delgada almidón gel método para enzima el mecanografiar de manchas de sangre". J forensic SCI. soc. 8 (2): 81 – 82. doi:10.1016/S0015-7368 (68) 70449-7. PMID5738223.
- ^ Buell, GN; Wickens, MP; Payvar, F; Schimke, RT (10 de abril de 1978). "Síntesis de cADN de la longitud total de mRNAs de oviducto parcialmente purificado cuatro". El diario de química biológica 253 (7): 2471 – 82. PMID632280.
- ^ ScHelp, C; Kaaden, o (mayo de 1989). «Mejora integral transcripción de ARN del virus de Sindbis por desnaturalización eficaz con metilmercurio hidróxido.». Acta virologica 33 (3): 297 – 302. PMID2570517.
- ^ Célula. 1979 Jan; 1:191-200. Separación independiente de la longitud de los fragmentos de restricción de DNA en electroforesis en gel bidimensional. Fischer SG, Lerman LS
- ^ Fromin N., J. Hamelin, S. Tarnawski, Roesti D., K. Jourdain Miserez, Forestier N., S. Cuvelle Teyssier, Gillet f el., M. Aragno y Rossi P. (2002) análisis estadístico de desnaturalizando electroforesis del gel (DGE) patrones de huella. E + nviron Microbiol 4:634-643.
- ^ Hempelmann E, RJ Wilson (1981). "Detección de la glucosa-6-fosfato deshidrogenasa en parásitos de la malaria". Parasitología molecular y bioquímica 2 (3 – 4): 197-204. doi:10.1016/0166-6851 (81) 90100-6. PMID7012616.
- ^ JR. Brody, Kern SE (octubre de 2004). "Historia y principios de los medios conductores para electroforesis de DNA estándar" (PDF). Anal. Biochem. 333 (1): 1 – 13. doi:10.1016/j.AB.2004.05.054. PMID15351274.
- ^ Lodish H; Berk A; Matsudaira P (2004). Biología celular molecular (5ª ed.). Freeman de WH: New York, NY. ISBN978-0-7167-4366-8.
- ^ Solución de electroforesis en gel de agarosa ADN. Enfoque p.66 19:3 (1997).
- ^ Thorne HV (1966). "Separación electroforética de polyoma virus ADN del ADN de la célula del anfitrión". Virología 29 (2): 234 – 9. doi:10.1016/0042-6822 (66) 90029-8. PMID4287545.
- ^ Weber, K; Osborn, m. (1969). "La fiabilidad de las determinaciones de peso molecular por dodecil sulfato-poliacrilamida gel electroforesis." El diario de química biológica 244 (16): 4406 – 12. PMID5806584.
- ^ Aaij C, Borst P (1972). "La electroforesis en gel de ADN". Acta de Biochim Biophys 269 (2): 192-200. doi:10.1016/0005-2787 (72) 90426-1. PMID5063906.
- ^ B Kastenholz (2004). "Electroforesis en gel de poliacrilamida continuo nativo preparatorio (PNC‐PAGE): un método eficaz para aislar cofactores de cadmio en los sistemas biológicos". Lett anal 37 (4): 657-665. doi:10.1081/AL-120029742.
- ^ Bier de Milán (ed.) (1959). Electroforesis. Teoría, métodos y aplicaciones (3ro impresión ed.). Academic Press. p. 225. OCLC1175404. LCC 59-7676.
- ^ Minde DP (2012). "Determinación de estabilidad biofísica de proteínas en lisados por un ensayo de proteólisis rápida, FASTpp". PLOS ONE 7 (10): e46147. doi:10.1371/journal.pone.0046147. PMC3463568. PMID23056252.
Enlaces externos
- Demostración de laboratorio Biotechniques electroforesis, del centro de aprendizaje de ciencia genética de la Universidad de Utah
- Electroforesis del gel de proteína nativa discontinua
- Electroforesis de paja de beber
- Cómo hacer un gel de DNA o RNA
- Animación de gel análisis de restricción del ADN
- Paso por las fotos de paso de correr un gel y extracción de ADN
- Un método típico de Wikiversidad
- Manual de métodos y principios de electroforesis en 2-D
|
||||||||||||||||||||||